
Sepmatix 8x平行色谱系统
助力药物合成产物筛选色谱柱
色谱应用
”
1
简介
药品属于一类化学或生物来源的产品,用于人类或动物的医疗。化学合成常用于药物的生产或潜在药物的开发。化学合成是一个经常与杂质存在有关的过程,因为收率很少是100%。这些杂质会对最终产品的功效、安全性和质量产生重大影响。因此对药物进行纯化以保证合成化合物的纯度和完整性至关重要,药物的纯化通常可以用色谱法进行。
近年来,超临界流体色谱(supercritical fluid chromatography,SFC)作为一种替代反相液相色谱(RP-LC)的方法出现了。SFC结合了气相色谱和液相色谱的优点。SFC使用超临界二氧化碳作为流动相的一部分,这是一种清洁和绿色的溶剂,很容易从最终产品中去除。此外,SFC提供了高分辨率和快速分离。
与RP-LC相比,C18柱对大多数样品至少达到可接受的选择性水平,在SFC中没有通用的通用固定相。在SFC中需要对不同的固定相进行筛选,以确定待分离样品的最佳选择性。二氧化碳的低极性允许非极性以及更多极性固定相整合到筛选过程中。确定最佳固定相并连续实施制备方法后,可对样品混合物进行纯化。
在本文中,合成了不同的药物并纯化它们。第一步,筛选不同的固定相,确定每种情况下的理想选择性。在第二步中,描述了用于实施制备纯化方法的过程。
2
实验设备
Sepiatec SFC-50
Sepmatix 8x SFC
PrepPure Silica, 5 um, 250 x 4.6 mm
PrepPure Diol, 5 um, 250 x 4.6 mm
PrepPure Amino, 5 um, 250 x 4.6 mm
PrepPure 2-EP, 5 um, 250 x 4.6 mm
HILIC, 5 um, 250 x 4.6 mm
PrepPure PEI, 5 um, 250 x 4.6 mm
PrepPure CBD, 5 um, 250 x 4.6 mm
Cyano, 5 um, 250 x 4.6 mm, (Dr. Maisch GmbH)
PrepPure 2-EP, 5 um, 250 x 10 mm
PrepPure PEI, 5 um, 250 x 10 mm
PrepPure Amino, 5 um, 250 x 10 mm
PrepPure Diol, 5 um, 250 x 10 mm
3
试剂和材料
普鲁卡因合成产物
二氧化碳 (99.9%)
甲醇 (≥ 99%)
2M氨气溶于甲醇中
甲酸 (99%)
去离子水
对乙酰氨基酚合成产物
利多卡因合成产物
乙酰水杨酸合成产物
苯佐卡因合成产物
为了安全操作,请注意所有相应的MSDS!
4
实验过程
运行条件 Sepmatix 8x SFC:
流动相:A =二氧化碳;B=甲醇
色谱柱尺寸:250 x 4.6 mm
流速:3ml /min(每根色谱柱)
流动相条件:0-0.5 min: 5% B
0.5-8.0 min:5-50% B
8.0-9.4 min:50% B
9.4-9.5 min:50-5% B
9.5-10 min:5% B
检测:200nm - 600nm 紫外扫描
筛选运行是完全自动开始的。使用流量控制单元将流量设置为每通道3ml /min,并平衡色谱柱。自动进样(V = 5 μL),开始平行筛选(运行时间= 10 min)。背压调节器设置为150 bar,柱箱加热至32°C。添加剂需要提前被添加到改性剂中。
SFC-50 运行条件:
流动相:A =二氧化碳;B=甲醇
柱尺寸:250 × 10mm
流动相条件:等度运行
检测:紫外
SFC柱在规定的流速下条件平衡3分钟。使用定量环自动进样,并开始运行。背压调节器设置为150 bar,柱箱加热至40°C。添加剂需要提前被添加到改性剂中。
5
实验结果
5.1 对乙酰氨基酚
对乙酰氨基酚(Acetaminophen,AA),也被称为扑热息痛,是一种镇痛、解热和非手性药物。它属于非阿片类镇痛药。化学上可以通过对氨基酚(AP)与乙酸酐反应合成,其中N-乙酰化发生(见图1)。为了确定对乙酰氨基酚合成产物纯化的理想选择性,第一步进行了色谱柱筛选(见图1)。
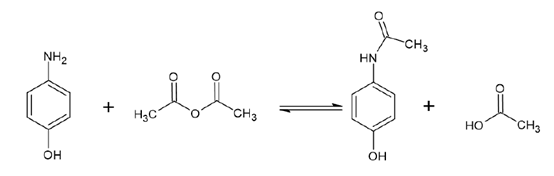

▲ 图1. 上:对乙酰氨基酚合成反应方程,下:筛选结果Sepmatix 8x SFC;从左到右色谱柱填料依次为:硅胶、氨基、二醇、氰基、2-EP、HILIC、PEI和CBD;运行时间= 10分钟。
共洗脱发生在二醇相和 2-EP 相。硅胶,CBD,氰基和氨基相没有表现出理想的选择性,因为没有基线分离可以在这里实现。HILIC和PEI相表现出良好的选择性和分辨率,因为分辨率总是高于1.5(见表1)。1.5的分辨率意味着两个峰可以很好地分离。表1 也显示了洗脱顺序。氰基相呈现出相反的洗脱趋势,对氨基酚先被洗脱,对乙酰氨基酚后被洗脱。筛选结果表明,该反应并非 100% 完成,产物中仍存在大量对氨基酚。
表1. SFC 条件下不同筛选柱的分辨率值和洗脱顺序
色谱柱 | R | 洗脱顺序 |
硅胶 | 0.95 | AA,AP |
氨基 | 0.58 | AA,AP |
氰基 | 1.62 | AP,AA |
二醇基 | X | X |
2-EP | X | X |
HILIC | 3.04 | AA,AP |
PEI | 5.57 | AA,AP |
CBD | 1.05 | AA,AP |
选择 PEI 色谱柱进行制备纯化,因为它具有最高的分辨率(见图2)。根据筛选得到的色谱图,利用梯度法可以确定 AA 和 AP 洗脱时流动相中甲醇的近似含量。该改性剂含量可作为等度制备方法的指导值。AA 和 AP 洗脱液中改性剂含量在 35-40% 之间。图2(上)为改性剂含量为 40% 时,在制备 PEI 柱上对AA的纯化。AA 和 AP 可以很好地分离。在相同条件下,可采用叠层进样法对 AA 进行自动化纯化(见下图2)。因为只有AA是相关的,所以不收集 AP。
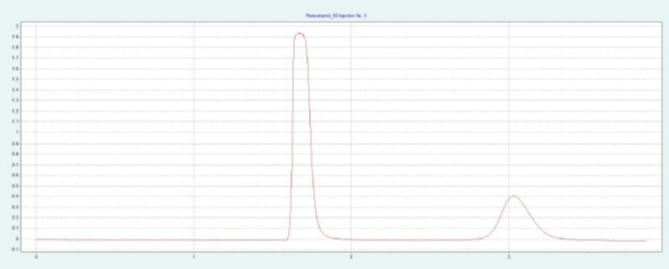
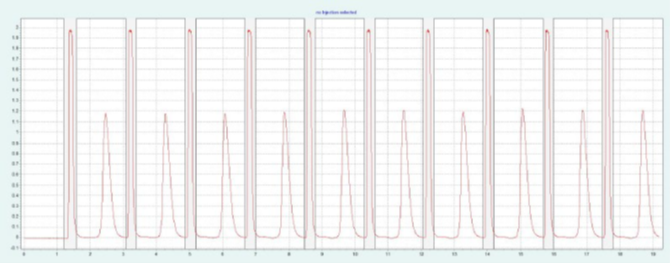
▲ 图2. AA纯化的单次进样(上)和叠层进样(下);运行条件:流量= 30 mL/min,改性剂为甲醇,改性剂% = 40%,温度= 40℃,背压调节器=150 bar,进样量= 250 μL,紫外波长= 254 nm;叠层进样条件:注射次数= 10次,叠层时间= 1.8 min,色谱峰收集 = 1(以时间为基础)。
5.2 利多卡因
利多卡因(L),化学式为2-二乙胺- N -(2,6-二甲基苯基)乙酰胺,是一种用作局部麻醉剂和抗心律失常药物的药物。它起到钠通道阻滞剂的作用。利多卡因可以两步合成(见图3)。在第一步中,2,6 二甲醚(X)的氨基被酰化。在第二步中,中间产物(IP)通过与二甲胺的亲核取代转化为利多卡因。因此,需要两个纯化步骤。色谱柱筛选的结果如 图2 所示。在改性剂甲醇中始终加入 20mm 氨水作为碱性添加剂。
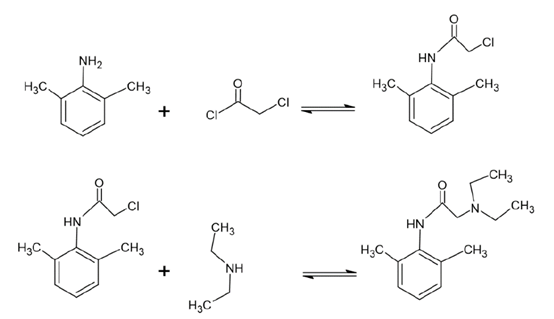

▲ 图3. 上:利多卡因合成反应方程,下:筛选结果Sepmatix 8x SFC;从左到右依次为:硅胶、氨基、二醇、氰基、2-EP、HILIC、PEI和CBD;运行时间= 10分钟
原则上,任何筛选的相都可以用于 IP 的纯化,因为总是可以实现基线分离。氨基相在改性剂含量相对较低的情况下具有最高的分辨率。X 和 IP 的洗脱顺序在每一相中都是恒定的。对于L的纯化,氰基和 CBD 相没有达到基线分离(见表2)。同样,氨基相显示出最高的分辨率。硅胶相是唯一表现出反洗脱趋势的相。否则,L 总是先洗脱。筛选结果表明,不可能每一步都达到 100% 的产率。这是不寻常的合成 IP,因为酸性氯化物是高活性的。据推测,在合成过程中,化学计量学出现了错误,导致 X 的添加过量。
表2. SFC 条件下不同筛选柱的分辨率值和洗脱顺序
色谱柱 | R1 | R2 | 洗脱顺序1 | 洗脱顺序2 |
硅胶 | 3.04 | 2.84 | X, IP | IP, L |
氨基 | 10.29 | 11.45 | X, IP | L, IP |
氰基 | 4.9 | 1.2 | X, IP | L, IP |
二醇基 | 3.35 | 3.44 | X, IP | L, IP |
2-EP | 4.18 | 5.67 | X, IP | L, IP |
HILIC | 7.63 | 6.33 | X, IP | L, IP |
PEI | 4.91 | 7.34 | X, IP | L, IP |
CBD | 2.81 | 1.12 | X, IP | L, IP |
选择氨基色谱柱进行制备性纯化,因为它具有最高的分辨率(见图4)。筛选结果表明,X 和 IP 的洗脱量约为 10-19%,L 和 IP 的洗脱量约为 11-19%。图4a 为改性剂 16% 时纯化 IP 的制备性单次进样,图4b 为改性剂 20% 时纯化L的制备性单次进样。在相同的条件下,可以进行叠层进样,分别对 IP 和 L 进行自动纯化(见 图4c 和 图4d)。因为只有 IP 或 L 是相关的,所以不收集反应物。
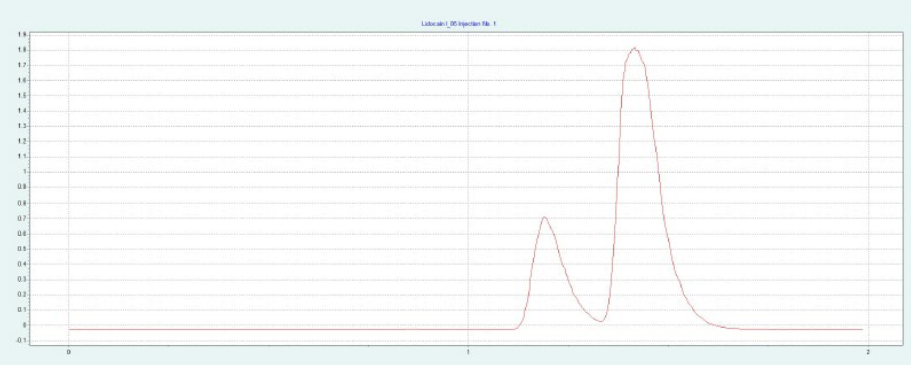
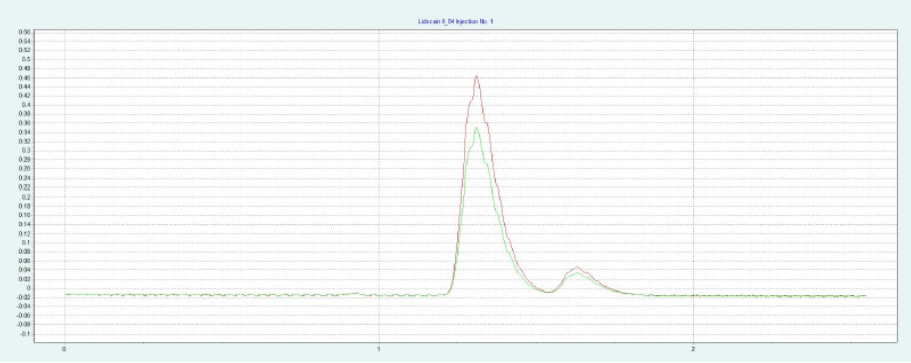
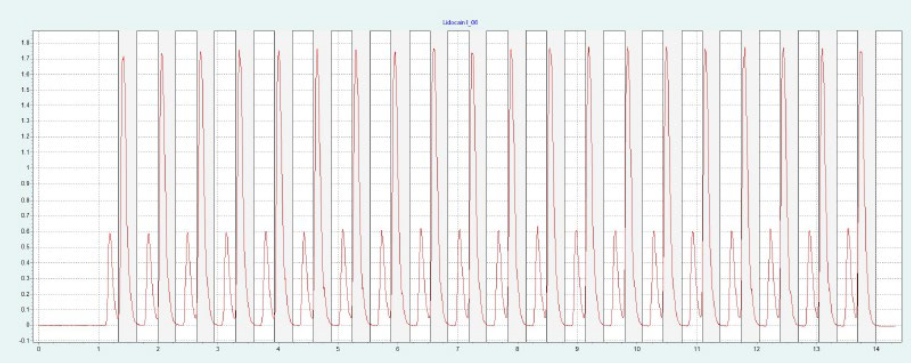
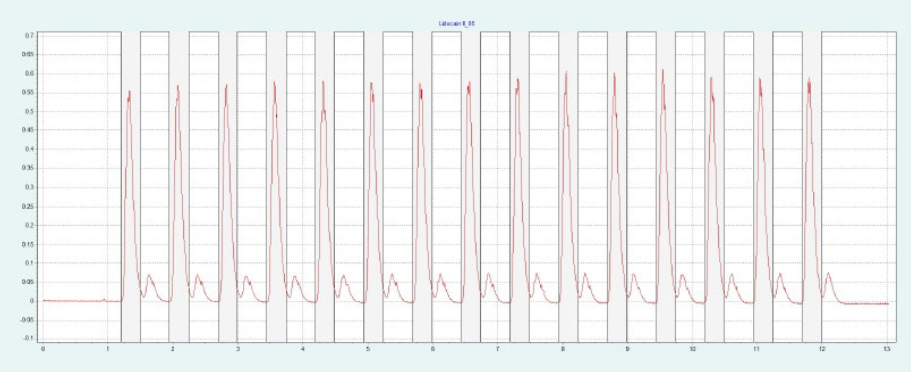
▲ 图4:单次进样(a)和叠层进样(c)纯化IP;运行条件:流量= 20 mL/min,改性剂=甲醇+ 20 mM氨,改性剂% = 16%,温度= 40℃,背压调节器=150 bar,进样量= 170 μL,紫外波长= 254 nm;叠层进样条件:注射次数= 15次,叠层时间= 0.75 min,色谱峰收集= 1(以时间为基础);L的纯化采用单次进样(b)和叠层进样(d);运行条件:流量= 20 mL/min,改性剂=甲醇+ 20 mM氨,改性剂% = 20%,温度= 40℃,背压调节器=150 bar,进样量= 170 μL,紫外波长= 254 nm;叠层进样条件:注射次数= 20次,叠层时间= 0.65 min,色谱峰收集= 1(以时间为基础)。
5.3 乙酰水杨酸
阿司匹林,化学成分为乙酰水杨酸(ASA),是一种众所周知的镇痛和抗炎药。它属于非甾体类抗风湿药。它可以通过水杨酸(SA)与乙酸酐的化学反应合成,并发生酯化反应(见图5)。为了确定乙酰水杨酸纯化的理想选择性,第一步进行了色谱柱筛选(见图5)。
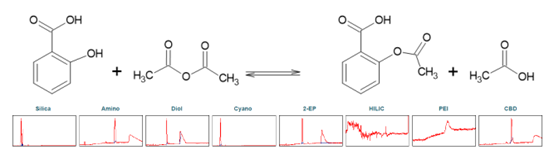
▲ 图5. 上:乙酰水杨酸合成反应方程,下:Sepmatix 8x SFC筛选结果;从左到右依次为:硅胶、氨基、二醇、氰基、2-EP、HILIC、PEI和CBD;运行时间= 15分钟。
由于 SA 在固定相中的高保留率,在梯度中添加 50% 改性剂 5 分钟的等度分离以促进 SA 的洗脱。HILIC 相只有噪声,ASA 和 SA 没有洗脱。在 PEI 相中,实验条件下只有 ASA 洗脱。除氰基相外,其余相均表现为 ASA 和 SA 的分离。2-EP 阶段显示了潜在运行时间和分辨率方面的最佳结果(参见表3)。筛选色谱图显示 ASA 合成没有达到 100% 产率。
表3. SFC 条件下不同筛选柱的分辨率值和洗脱顺序
色谱柱 | R | 洗脱顺序 |
硅胶 | 1.68 | SA, ASA |
氨基 | 1.79 | ASA, SA |
氰基 | X | X |
二醇基 | 3.86 | ASA, SA |
2-EP | 6.85 | ASA, SA |
HILIC | X | X |
PEI | X | ASA |
CBD | 27.45 | ASA, SA |
选择 2-EP 相进行制备纯化。筛选结果表明,改性剂含量约为 26-40% 时,ASA 和 SA 可被洗脱。图6(上)为改性剂含量为 40% 时,ASA 在 2-EP 制备柱上的纯化结果。在改性剂中加入 0.5% 甲酸,改善 SA 的峰形。SA 在没有甲酸的情况下显示出很强的尾砂。随着甲酸的加入,SA 的尾砂峰值降低。在相同条件下,可以采用叠层进样法对 ASA 进行自动化纯化(见下图6)。因为只有 ASA 是相关的,所以不收集 SA 和其他杂质。
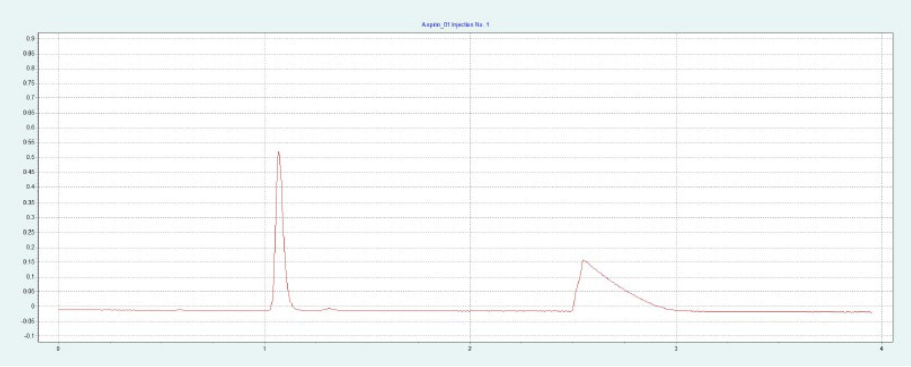
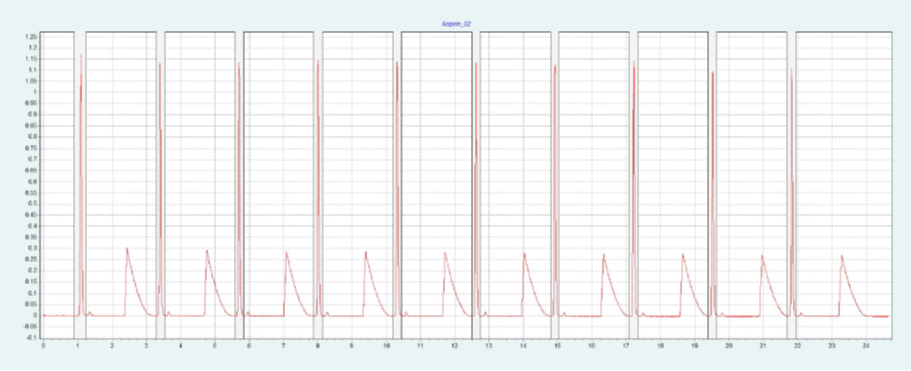
▲ 图6. ASA纯化的单次进样(上)和叠层进样(下);运行条件:流速= 30 mL/min,改性剂=甲醇+ 0.5%甲酸,改性剂% = 40%,温度= 40℃,背压调节阀 = 150 bar,进样量= 200 μL,紫外波长= 254 nm;叠层进样条件:注射次数= 10次,叠层时间= 2.0 min,色谱峰收集= 1(以时间为基础)。
5.4 苯佐卡因
苯佐卡因(BC),化学名称为 4-氨基苯甲酸乙酯,属于局部麻醉剂。以对氨基苯甲酸(PABA)为原料,羧基与乙醇经酸催化酯化反应可合成对氨基苯甲酸(PABA)。硫酸被用作催化剂。进行柱筛选以确定苯佐卡因合成产物纯化的理想选择性(见图7)。
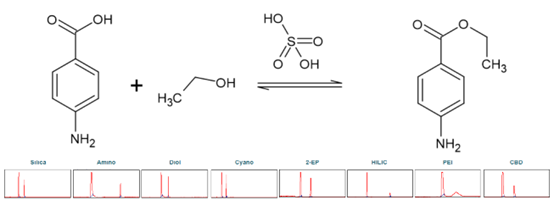
▲ 图7. 上:苯佐卡因合成反应方程,下:筛选结果Sepmatix 8x SFC;从左到右依次为:硅胶、氨基、二醇、氰基、2-EP、HILIC、PEI和CBD;运行时间= 15分钟。
由于PABA在固定相中的高保留率,在梯度中增加了一个在50%改性剂下5分钟的等度步骤。由于酸性,用洗涤步骤除去了硫酸。各固定相的色谱图表明,各固定相均能分离苯佐卡因。在洗脱速度和分辨率上存在较大差异(见表4)。苯佐卡因总是在PABA之前被洗脱。HILIC相和氨基相的分辨率最高,而PABA相的保留率很高,即使改性剂含量达到50%。色谱图也表明酯化反应尚未完成。
表4. SFC 条件下不同筛选柱的分辨率值和洗脱顺序
色谱柱 | R | 洗脱顺序 |
硅胶 | 6.38 | BC, PABA |
氨基 | 21.6 | BC, PABA |
氰基 | 5.38 | BC, PABA |
二醇基 | 10.82 | BC, PABA |
2-EP | 10.86 | BC, PABA |
HILIC | 28.78 | BC, PABA |
PEI | 1.94 | BC, PABA |
CBD | 8.72 | BC, PABA |
选择二醇相进行制备纯化。HILIC 相和氨基相要求改性剂含量高,运行时间长。二醇相显示样品的快速洗脱,具有足够高的分辨率。筛选结果表明,改性剂含量约为 30-40% 时,BC 和 PABA 均可洗脱。图8(上)显示了在改性剂含量为 28% 的制备二醇柱上纯化 BC。在相同条件下,可以采用叠层进样法对 BC 进行自动化纯化(见下图8)。因为只有 BC 是相关的,所以不收集 PABA。
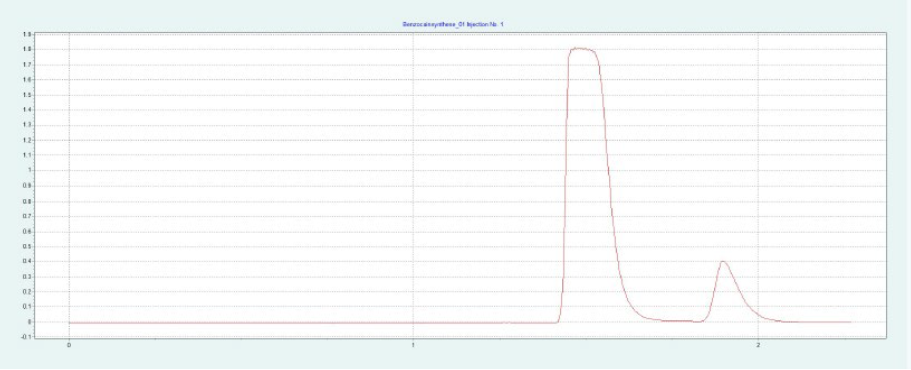
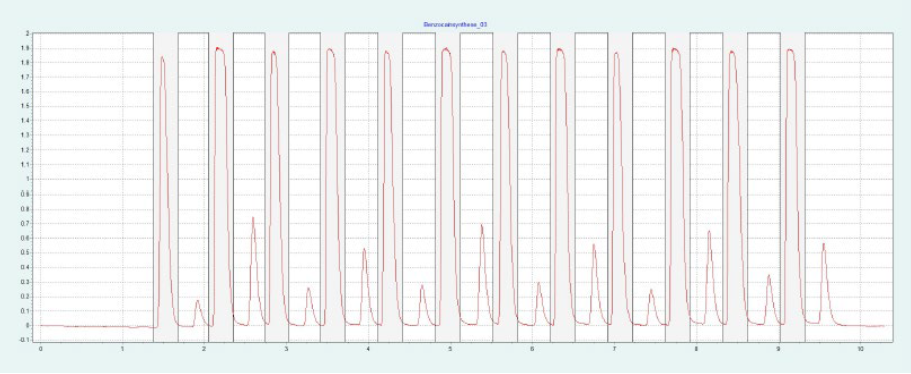
▲ 图8. 苯佐卡因纯化的单次进样(上)和叠层进样(下);运行条件:流速= 20 mL/min,改性剂为甲醇,改性剂% = 28%,温度= 40℃,背压调节器 = 150 bar,进样量= 90 μL,紫外波长= 276 nm;叠层进样条件:注射次数为12次,叠层时间为0.66 min,色谱峰收集=1(以时间为基础)。
5.5 普鲁卡因
普鲁卡因(PC),化学式为4-氨基苯甲酸2-(N,N-二乙胺)乙酯,是一种药物,用作局部麻醉剂。它阻断了依赖电压的钠离子通道,从而导致对疼痛的敏感性降低。普鲁卡因是由4-氨基苯甲酸乙酯(ABE)化学碱催化合成的。这涉及到与2-二乙胺乙醇的酯交换反应。该碱为乙醇酸钠,可由单质钠与乙醇反应生成。进行柱筛选以确定纯化普鲁卡因合成产物的理想选择性(见图9)。
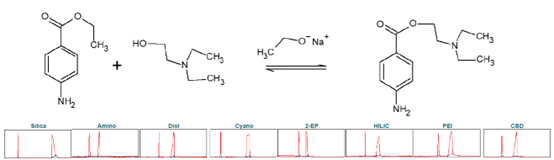
▲ 图9. 上:普鲁卡因合成反应方程,下:Sepmatix 8x SFC筛选结果;从左到右依次为:硅胶、氨基、二醇、氰基、2-EP、HILIC、PEI和CBD;运行时间= 15分钟。
由于 PC 在固定相中的高保留率,在梯度中增加了一个在 50% 改性剂下 5 分钟的等度步骤。原则上,每个固定相对普鲁卡因的纯化都有合适的选择性(见表5)。普鲁卡因在硅胶、PEI和氰基相中具有较高的保留率。由于普鲁卡因的基本性质,在筛选条件下,相应峰的峰形较差。二醇相为强前向,不对称度为0.58;硅胶相为强尾向,不对称度为 2.73。ABE 后每个固定相的 PC 洗脱。
表5. SFC 条件下不同筛选柱的分辨率值和洗脱顺序。
色谱柱 | R | 洗脱顺序 |
硅胶 | 16.65 | ABE, PC |
氨基 | 6.54 | ABE, PC |
氰基 | 8.59 | ABE, PC |
二醇基 | 9.16 | ABE, PC |
2-EP | 5.29 | ABE, PC |
HILIC | 5.67 | ABE, PC |
PEI | 3.69 | ABE, PC |
CBD | 7.26 | ABE, PC |
选择 2-EP 相进行制备纯化。硅胶相需要高含量的改性剂,这导致运行时间长,峰形差。2-EP 相显示样品的快速洗脱,具有足够高的分辨率。甲醇改性剂中加入 5% 去离子水和 20mm 氨水作为添加剂。筛选结果表明,改性剂含量为 24 ~ 32% 时, BC 和 PABA 的洗脱效果较好。图10(上)为改性剂含量为 25% 时,PC 在 2-EP 制备柱上的纯化结果。在相同条件下,可采用叠层进样法对 PC 进行自动化纯化(见下图10)。添加剂的加入显著改善了 PC 的峰形。
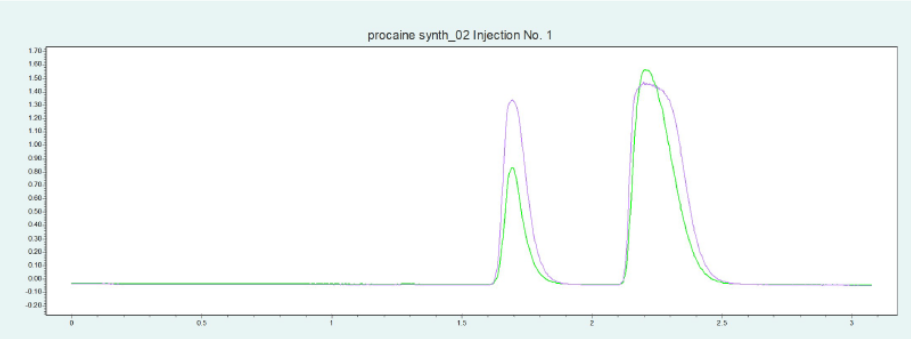
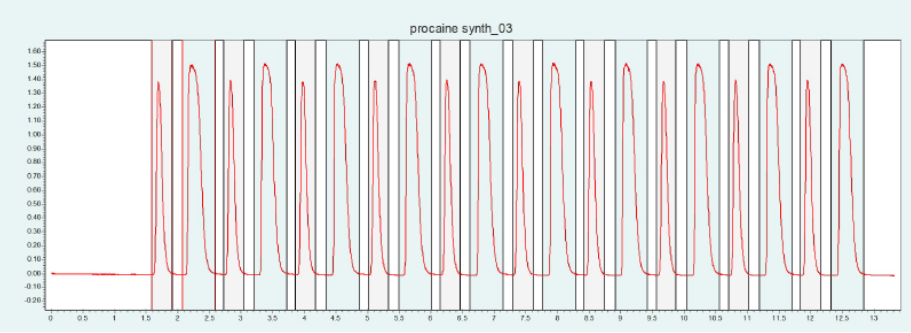
▲ 图10. 普鲁卡因纯化的单次进样(上)和叠层进样(下);运行条件:流量= 20 mL/min,改性剂=甲醇/水(95/ 5%),含20 mm氨水,改性剂% = 25%,温度= 40℃,背压调节器 = 150 bar,进样量= 100 μL,紫外波长= 220 nm;叠层进样条件:注射次数= 10次,叠层时间= 1.1 min,色谱峰收集= 2(以时间为基础)。
6
实验结论
在进行有机合成后,由于副反应或转化产物收率不是 100%,杂质通常存在。这些杂质必须除去,特别是在药品生产中。在本文中,使用 Sepiatec SFC-50 仪器纯化了几种药物。药物合成后,使用 Sepmatix 8x SFC 仪器对每种药物进行色谱柱筛选,以确定纯化工艺的最佳色谱柱。最后采用最合适的色谱柱进行制备。合成产物的柱筛选表明,不同的固定相往往具有最佳的纯化选择性。因此,在纯化前确定最合适的色谱柱是非常重要的。如实例所示,筛选结果可用于实施一种简单的制备方法。如果合成混合物不是太复杂,可以使用叠层进样进行以节约纯化时间。
